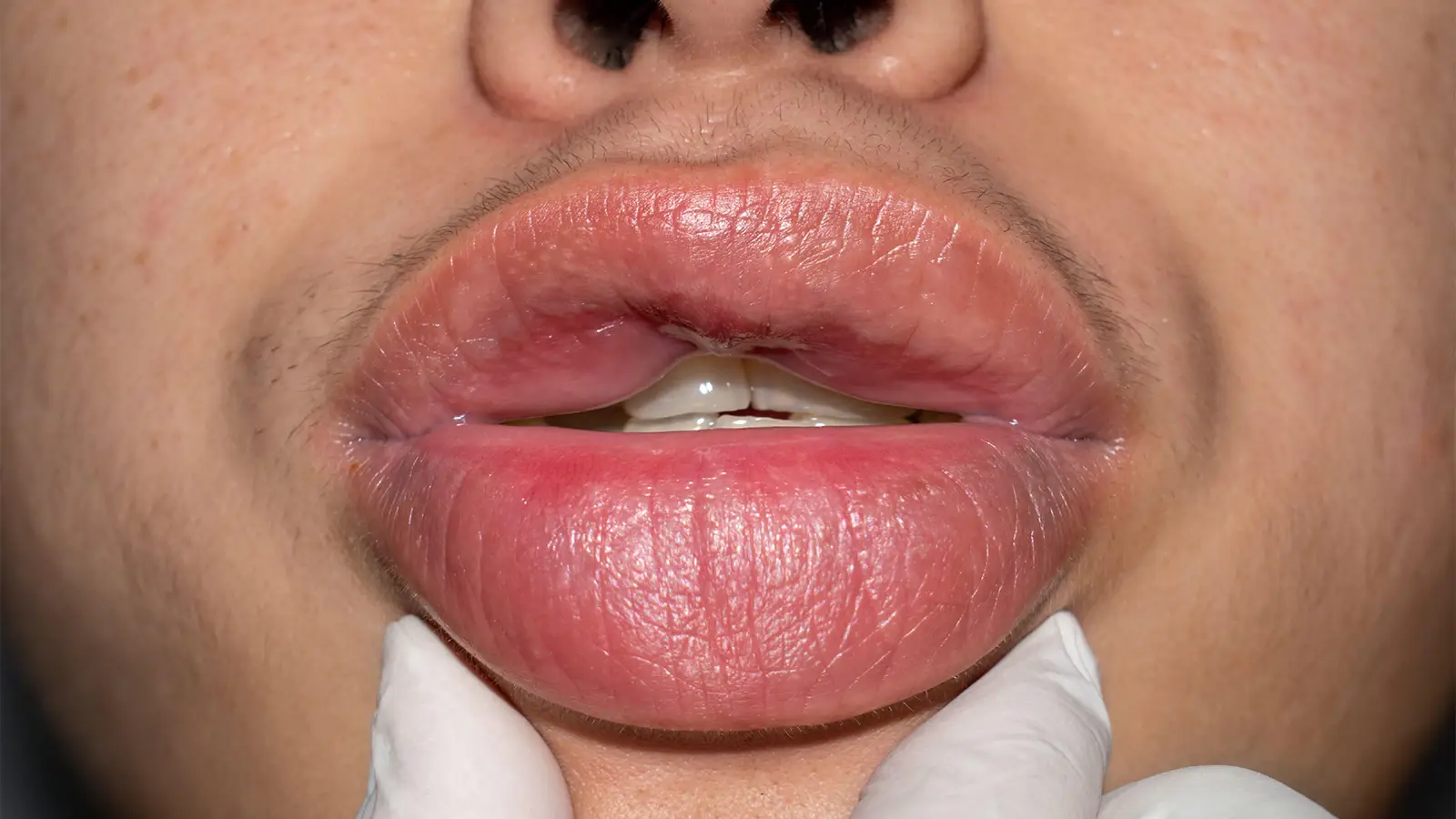
Hereditary angioedema (HAE) is a rare autosomal dominant genetic disorder related to low levels of C1 inhibitor, which lead to too much of the plasma kallikrein enzyme, triggering overproduction of bradykinin that binds to receptors on blood vessel walls that then dilate and allow fluid accumulation in surrounding tissues.
This temporary localized increase in vascular permeability produces the characteristic swelling of subcutaneous, mucosal, and submucosal tissue without associated pruritus or wheals. These attacks primarily impact the skin of the extremities, trunk, and face, as well as the gastrointestinal tract, genitals, and larynx. When the condition affects the larynx, which it does in half of patients, fatal asphyxiation due to obstruction of the upper airways is a major risk.
HAE with C1 inhibitor deficiency, characterized by reduced functional levels of C1 inhibitor, is the more common form. It often starts early in life, with 50% of patients showing symptoms by age 10 years that escalate in frequency and severity after puberty, and continue throughout adulthood. The type 1 subtype accounts for 85% of these cases and is associated with low antigenic C1 inhibitor protein levels; type 2 has normal antigenic levels. Both have been linked to SERPING1 genes, which encode C1 inhibitor, and are clinically similar.
In 2000, HAE with normal C1 inhibitor was first described. This form with quantitatively and functionally normal C1 inhibitor levels has been linked to mutations in the F12 gene, although it is still less understood than HAE with C1 inhibitor deficiency. Symptoms typically begin in late teenage or early adult years.
The U.S. Hereditary Angioedema Association Medical Advisory Board guidelines note that “angioedema symptoms may affect the face and tongue more frequently in [HAE with normal C1 inhibitor] with fewer abdominal symptoms compared with [HAE with C1 inhibitor deficiency].”
Other differences include lower and variable penetrance of HAE with normal C1 inhibitor in families. Females with HAE with normal C1 inhibitor are more likely to be symptomatic than males, “and the swelling is often estrogen-sensitive such that exposure to endogenous or exogenous estrogens is a strong exacerbating factor,” the guidelines state, although rare cases have occurred in males.
While the prevalence of HAE with normal C1 inhibitor has been estimated to be much lower than the other forms, ICD-10 diagnostic codes used in the U.S. don’t have any specifically for HAE, making estimates less straightforward. However, some recent data attempted to provide a more accurate picture for the U.S. population.
A 2025 analysis of IQVIA’s PharMetrics Plus MedTech claims database using codes for “angioneurotic edema” or “defects in the complement system” plus at least one pharmacy claim for a HAE-indicated medication as a surrogate for the condition showed an overall annual unadjusted prevalence of 2.43 per 100,000 persons with any type of HAE in the most recent year (2020). With physician review of the anonymized claims data, the estimate dropped to 1.84 per 100,000, or 6,595 persons in 2020.
Prior frequently cited numbers for HAE with C1 inhibitor deficiency have been in the 1 to 2 per 100,000 persons range.
A 2023 survey of clinicians in the Hereditary Angioedema Association estimated that 1,230 to 1,331 patients had HAE with normal C1 inhibitor, for a prevalence of 0.37 per 100,000.
Without biomarkers for these conditions, diagnosis of HAE largely rests on clinical criteria. Untreated, angioedema symptoms commonly last 3 to 5 days but not weeks or longer. Another important clue to diagnosis is that symptoms do not respond to antihistamines, corticosteroids, or epinephrine.
Guidelines recommend that appropriate testing includes measurement of serum C4 levels, C1 inhibitor antigenic levels, and C1 inhibitor functional levels. Low C4 plus low C1 inhibitor antigenic or functional levels can help diagnose HAE with C1 inhibitor deficiency. Symptoms plus normal C1 inhibitor tests should prompt additional genetic tests for factor XII, plasminogen, angiopoietin-1, and kininogen mutations when available to diagnose HAE with normal C1 inhibitor.
“Targeted next-generation sequencing and novel biomarkers may soon improve the diagnostic approach for [HAE with normal C1 inhibitor],” the guidelines note. “Considering how dramatically HAE care has changed over the past decade, there is considerable enthusiasm that many of the extant problems will be solved in the near future.”
Source link : https://www.medpagetoday.com/spotlight/hae/121072
Author :
Publish date : 2026-05-01 17:41:00
Copyright for syndicated content belongs to the linked Source.








{kind=link}